

Aspirations of Biological Monosemanticity
Aspirations of Biological Monosemanticity
How to Move Past Target-Based Drug Discovery and Realize the Future of Precision Medicine

Over the course of the past twenty years, there has been a lingering promise of molecular medicine, born of the Human Genome Project, that has gone largely unfulfilled. The tools available to the modern biological scientist dwarf the capacity of our predecessors by multiple orders of magnitude, as does the organized core of information that we all possess and can access with basic internet service. And yet, the pharmaceutical industry has experienced a steady decline in productivity despite increasing researcher workforces. The failure rate for drugs in the clinic is 90%, with the majority of these clinical compounds showing a lack of efficacy or an intolerable toxicity. The promises of solving medicine using molecular techniques seem increasingly far away, and the biomolecular dreams of the early aughts have been hardened by reality. Any physician, scientist, or medical expert will decry the quality of research that now exists, and the subsequent need for meta-analyses and scientific consensus built upon shaky foundations. The biological replicability crisis and the declining productivity of the biotechnology industry are not only fundamentally intertwined, but they are symptoms of the same scientific philosophy that has plagued medical research. Not only are we capable of surpassing this philosophical stagnancy and delivering a future that can harness a true mastery of our cellular biology, but the mechanisms by which we can overcome these failures are being built, independently, as we speak.
Let’s start from the beginning.
What is Target-Based Drug Discovery?
The current dominant philosophy in drug discovery pursuits in both academia and industry is known as target-based drug discovery (DD). This paradigm involves, at a simplistic level, the identification of a novel molecular target implicated in disease, the selective chemical or biological modulation of that target, and the adaptation of the learned chemical or biological matter for human physiology. This is a complex process that lasts years over the course of continuing efforts, often through both academic and industry research. Targets are proteins, identified through biological screens or through association with a known disease state. Models are created to show that the inhibition or degradation of that protein results in an alleviation of markers of that disease state (Step 1). Chemical matter can then be designed to selectively inhibit that target over all others (Step 2), and that chemical matter can then be refined to minimize pharmacokinetic / pharmacodynamic (PKPD) issues in the human body (Step 3). This design process came about during the biochemical revolution of the late 20th century, during which the molecular mechanisms of disease were discovered for the very first time. Since then, the individual steps of this process have been optimized in many ways, but the fundamental philosophy of design has changed very little.

Technology has adapted and evolved to serve this specific design ideology. AlphaFold, the Artificial Intelligence darling of the structural biology community, has enabled computational folding of proteins to determine structure, accelerating the development of rationally designed ligands. My field of expertise, high-resolution mass spectrometry, has allowed for fast parallel processing of the entire proteome to determine target selectivity of a new ligand. Both of these fields have experienced extraordinary gains in the 2nd step of drug discovery labeled above, using the most cutting edge technology that humankind has to offer. At the center of it all is the idea of the target protein. Every portfolio project in my research division is named by its target. We as medical researchers think, and understand the world in terms of target proteins. These advances are built upon the assumption that if we understand thoroughly enough the target protein we seek to inhibit, and inhibit that target and that target alone, we will have a safe and effective drug. This assumption is false.
Neither of these amazing advances does anything at all to to solve the fundamental problem of target-based drug discovery, which exists in Step 1 and before. To understand why target-based approaches fall and will always fall short of our biochemical aspirations, we need to start at the beginning one more time.
What existed before Target-based DD?
According to a 2023 systematic review of 150 years worth of articles and patents, only 9.4% of all approved drugs were discovered using target-based approaches, despite more than two decades of ideological dominance in the most technologically advanced period in history. Even amongst those 9.4%, there is almost certainly a degree of off-target action that supports the promoted phenotypic efficacy. A 2019 review found that for multiple clinical candidates for cancer maintain their efficacy even when the protein that they supposedly target, that the drug development process was built upon, was knocked out using CRISPR. This is such a groundbreaking result that it bears repeating. These are drugs that patients are receiving, that undergo completely different mechanisms than all scientists involved specifically designed them for. Physicians are taught biology that is completely false, and scientists are basing their expertise on the same. How, then, did these compounds show efficacy and as such were progressed to the clinic? The answer lies in the same way that drugs were developed before molecular biology.
The first paradigm of drug discovery is indigenous knowledge; that is, people using a basic combination of trial-and-error and word-of-mouth knowledge. The ancient Sumerians discovered that eating the willow leaf would cure pain, and today we have aspirin. Europeans in the 18th century discovered that eating lemons and oranges would cure scurvy, and today we have Vitamin C. So on, and so forth.

The second paradigm is a closely related, more organized and accelerated version of paradigm one known as phenotypic drug discovery. In phenotypic drug discovery, a specific phenotype is selected for using chemical matter. For example, a screen may be created to look for molecules that specifically inhibit the growth of a bacteria or fungus. These hits can then be selected against safety screens to make sure human biology is not inhibited, and a drug is born. Phenotypic drug discovery is the source of the majority of currently approved drugs, and dodges most of the need for target-based enhancements that exist in target-based DD. In fact, many successful drugs discovered through phenotypic screens often do not have a single high-potency target — they bind to many different targets with varying degrees of potency. The immediate downside to phenotypic drug discovery, however, lies in the issues of scalability and superficial knowledge that are also present in paradigm 1. It may be difficult to design a sufficiently complex screen for a phenotypic result given the necessary complexity of chemical matter. Furthermore, there are very real threats to maintaining a superficial solution to a complex biological problem, one of which was illustrated by the aforementioned discovery of vitamin C.
In the 18th century, scurvy was a major killer on long transoceanic voyages. The discovery of citrus juice as a cure was a major breakthrough that saved countless lives of sailors on their journeys. However, with the development of the steam engine, transoceanic voyages were shortened to the point where scurvy was no longer a concern. Citrus juice manufactures switched from lemons to limes, which despite having half the vitamin C were cheaper to produce in the Mediterranean climate. They also began to heat the juice for preservation purposes, which significantly broke down the remaining vitamin C. However, given the shortened transatlantic journeys, these effects went unnoticed. With the advent of the Antarctic exploration crews, journeys were suddenly much longer, and sailors quickly realized that their preserved lime juice was no longer enough to fend off scurvy. The superficiality of the solution was revealed, and people lost their lives to a disease that had already been fundamentally “solved”.
Phenotypic drug discovery has its advantages, but its disadvantages were quickly overcome by the third paradigm of drug discovery, target-based. In target-based DD, we could use our newfound knowledge of molecular biology to fundamentally solve a problem once and for all, though it required a more thorough discovery process. I believe it is possible to combine the advantages of both phenotypic drug discovery and target-based drug discovery to yield a new, fourth paradigm of medical investigation. Under this philosophy, scientists would use complex holistic phenotype-adjacent models while developing a reductionist, biophysical understanding of the mechanisms of disease. Scientific investigation could progress beyond simple, low-hanging fruit of easy targets and monogenic diseases and pursue complex biological networks while understanding the fundamental physics of our medical interventions. This paradigm may be closer than we think, and the tools to create it are evolving as we speak.
AI and implications for Drug Discovery
In a 2023 analysis of the use of Artificial Intelligence in drug discovery, the authors describe the nascent field of AI DD, and where AI has most effectively been used. As is the case with AlphaFold, the majority of AI advances appear to be implemented in the design of molecular properties of drugs, antibody affinity binding, and specific tasks. This makes general sense — the easiest way to implement AI into the existing framework of drug discovery is to use it for specific concrete tasks, where an AI can be trained on existing data and give a common output, such as affinity, or toxicology, or ligand binding. These computational tools are useful, but again serve to perpetuate the framework of target-based drug discovery. How can we begin to use AI to fundamentally integrate phenotypic with target-based drug discovery?

Pictured above is a simple neural network. The artificial neural network is a type of AI, inspired by the structure of the brain, that lies at the heart of the most complex AI systems being developed right now. The network receives a number of inputs (blue circles) which transmit a set of signals to the hidden layers (black circles). These hidden layers pass the signal along until the outputs (green circles) receive a signal to present. These models are trained by adjusting the relationship of each of the nodes (the circles) to each other in order to minimize the error between the predicted output from the green nodes and the actual output. In the case of ChatGPT, the input is a set of text and the output is the prediction of the next word. In the case of your brain, the input is all of your sensory data + a top-down prediction of what the world should be and the output is your lived experience (This is vastly simplified). Using these convolutional learning patterns, an artificial neural network can learn to solve complex problems. This can be thought of even more simply in terms of the most basic machine learning function — the linear regression.
In linear regression, there are two nodes. Input (blue) represents the X value, also known as a feature. Output (green) represents the Y value. This relationship is stored in the form:
y = mx + b
That you may all be familiar with. During training, each data point in the training data informs what the weight (m) and the bias (b) should be in the relationship in order to best fit the trendline to the data points. Afterwards, we are left with a compressed version of the dataset that is now stored in two specific points, m and b. These points allow us to do two things: first, we can predict what an output should be, approximately, given a specific input. Second, the model itself contains information about the relationship of the input and output. If the slope of the line is 2, then for every 1 that our X value increases, our Y value should increase approximately two. We can describe the relationship between X and Y.

Looking at our neural network compared to a linear regression, we begin to appreciate its complexity. We have a large network of nodes, each of which maintains a relationship to multiple other nodes that is uniquely tuned to that specific relationship. We can use a trained neural network to predict an output given a new input, and the neural network also contains information about the fundamental relationships of the system within its nodes. When Google released its Transformer network architecture in 2017, the model displayed incredible capabilities in next-word-prediction tasks — and surprisingly, the model also possessed the ability to translate languages with very little prompting. The model doesn’t just predict the next word, it encodes the fundamental rules of the system. In this case: linguistics.
The AI Interpretability Problem
The problem with deep learning is that the inside of a neural network is effectively a black box. Researchers train an AI exclusively using the inputs and outputs, without viewing what occurs in the hidden layers. The relationships between hidden layers naturally develop in pursuit of minimizing prediction error between the predicted output and the true output. This concern has birthed the whole field of AI Safety, wherein experts aim to understand the inner workings of an AI the way that neuroscientists attempt to interpret the brain. Specifically, Large Language Models (LLMs) like ChatGPT frequently give strange, incorrect, patronizing, or vaguely threatening answers, and researchers would like to deconvolute what exactly is occurring within the neural network that leads to these aberrant responses. Approximately two months ago now, leading AI industrial lab Anthropic released a new paper on the AI interpretability problem that may glean some important insights.
In Anthropic’s Towards Monosemanticity, researchers seek to understand the ways in which a neural network encodes information within its hidden layers. In an ideal world, there would be a node that encodes the word “biology“ and therefore turns on specifically for concepts related to biology. This phenomenon, known as monosemanticity, is sometimes the case, but is very rare. For the majority of nodes within a network, each appears to be polysemantic, activating on a variety of unrelated topics, whether that be biology, trains, oranges, or past participles in Portuguese. The researchers describe this phenomenon in a separate paper as a sort of superposition: individual nodes within the network compress multiple ideas into the relationship between multiple nodes.
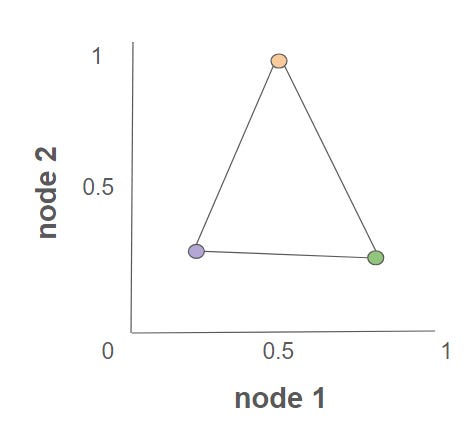
For example: a triangle compression would compress three ideas (features) into two dimensions (nodes). If node 1 is 0.5 and node 2 is 1, the network is passing ORANGE. If node 1 is 0.8 and node 2 is 0.2, then the network is passing GREEN. As Anthropic trains a small AI on text prediction, the AI experiences node superposition that increases in feature:dimension ratio as more information is encoded into a set number of nodes.

Furthermore, the team realizes that these superimposed shapes can be reversed, by training a larger AI to predict the individual behavior of the nodes of a smaller AI that has been trained to predict text. While the behavior of the small model is polysemantic due to a high degree of superposition, the large model trained on the small model is largely monosemantic.

Node #3768 of the large model fires on “kinase”, and is highly correlated with neurons that fire on medical and biochemical terminology. The large model interprets the fundamental characteristics encoded in the small system, mapped out in a way that’s observable. Polysemanticity of the natural network is effectively reduced down to a model we can understand, and modulate.
The 4th Paradigm: Convolutional Drug Discovery
Human biology exists in-cell as an unstructured convolutional network. Each protein impacts all other proteins to various degrees depending on their folding and physical properties, and many proteins in turn affect the overall condition of the cell (i.e. serve as output nodes).

The failure of target-based drug discovery is fundamental to its philosophy: proteins are nodes, not features. They are not themselves the signal, they are the medium through which the signal travels. Like a soundwave through air molecules, or the concept of a word through an artificial neural network, or a thought through your mind — disease is caused fundamentally by aberrations of signals passing through proteins. Proteins, like nodes in a neural network, are highly polysemantic. Each individual protein in the proteome has always seemingly been implicated in a variety of different systems, from metabolism to trafficking to homeostasis. This is due to the fact that each protein, like nodes in a neural network, experiences the same superposition of signals in relation to other proteins in the proteome. It is these signals, not the proteins themselves, that ought to be targeted in drug discovery efforts.
In the case of growth signal cascades in cancer, the issue is not fundamentally with CDK4. It is not with KRAS, or PI3K, or any other protein that we seek to modulate. It is with the signal that is constitutively active, that must be restored to biological normal. If a network is dysfunctional, the logical solution is hardly to fully inhibit a node with maximum potency, or degrade a node with maximum specificity. In the future I imagine the biochemists will view our approach of degrading or inhibiting targets with high potency as we view the lobotomists of the 20th century. Each displays a desperate lack of knowledge of the complexity of the system at hand and so relies upon brute techniques for modulation.
These issues are worsened by the fact that modern medical research has been building these networks over the past 80 years one edge at a time, using old techniques that modern science has largely proven to be inaccurate. These networks as we understand them are highly inaccurate, and more complicated than we imagine. There are thousands of proteins within the convolutional network, before even a single consideration of local microstructures like organelles and cellular receptors. Our understanding of biological pathways is largely inaccurate and certainly incomplete. It is fortunate, then, that our current pursuit of AI interpretability is perfectly positioned to illustrate complex, obscure networks.
How to revolutionize modern medicine
Just as the researchers at Anthropic prove that it is possible to deconvolute the superposition of a polysemantic node using a larger convolutional model, it ought to be possible to deconvolute the features being passed through the unstructured network of human biochemistry using a larger, more powerful model. In this regard, deconvoluting biological models appears to be much easier than deconvoluting a frontier AI model. In Towards Monosemanticity, the researchers achieve successful deconvolution of a 512 single-layer neuron model using models with 256x the neuron count. Achieving such a large encoder model on GPT-4, with its rumored billions of neurons, seems much more unmanageable than the human proteome with ~17k proteins and ~51k phosphosites.

To accurately map the convolutional network of human biochemistry, then, would require a large AI model that could be trained on the existing library of multi-omics data. Such a model, trained to predict the abundance or phosphorylation of a protein given the abundances and phosphorylations of the remaining proteome, could decode the relationships that are foundationally embedded in human biochemistry, some of which we have mapped already. The individual nodes of the large model, then, would represent the features of biochemistry. These are the signals that are being passed through proteins in order to differentiate health and disease, and it is these signals that ought to be the focus of our attention as an industry. Then, in order to quiet or activate an offending signal, there is no longer a need to target and inhibit a specific node within that pathway, a node that is likely polysemantic to a whole host of other evolutionarily conserved functions. Drug discovery efforts in the future could be focused on targeting the edges of the bionetwork, known as protein-protein interactions (PPIs). By enhancing or discouraging certain PPIs within a network, specific relationships and therefore signals could be curbed while minimizing off-feature toxicity. These drugs could have lower potencies and be easier to tolerate, and taken in cocktails to affect multiple edges within one signal pathway.
This will require not just a momentous bioinformatic effort but also an industry-wide shift in philosophy, practices, and portfolios, all of which possess considerable inertia. However, too much is at stake not to capture the 90% of drugs that fail. This is the problem of our generation, and the solution will effectively accelerate modern medicine to deliver better, faster, cheaper cures to the patients to need it the most.
This is great. I love the part where you describe that molecular/cellular/systems pathways are the MEDIUM through which signals are transmitted, and not the signals themselves.
When I read Scott's summary of 'toward monosemanticity' last week I thought the same thing about the longstanding problem of interpretability in biological signaling cascades. You read so many papers that say, "mechanism X is dependent on NFkappaB" or some such nonsense, as though that's meaningful to anyone. Any given Tuesday is likely to induce NFkappaB signaling. Same with the thousands of 'signaling cascades in [cancer/immunity/cardiology/metabolism/insert field here]' posters hanging around academic centers. So much wasted paper for a bunch of scientists to run around pretending they understand something they're all ignorant about. Don't flex when you don't have the muscles for it.
Until now we've simply not had a good explanation for how a single input is converted by the multiplicity of signals passing through cellular systems into the range of outputs we observe. The one-signal/one-output model has been obviously false for a long time - no matter how you modify it. Yet as you noted in your post, "Physicians are taught biology that is completely false, and scientists are basing their expertise on the same." I feel like that statement should become a disclaimer footnote on every poster/slide/paper that includes a cell signaling cascade diagram.
Now, I'll say three things about converting polysemantic signals (alternately signals in superposition) into monosemantic models. (Sorry this comment is so long.)
First is that when I read Scott's post on this last week I immediately thought of this longstanding problem in biology and it felt like the same mind-blowing moment as when I first learned about protein folding and how that leads to all the observable properties of the biological world. The polysemanticity -> monosemanticity approach finally felt like we had a Rosetta Stone we could use to read the impossible languages of biology. It was like, "How do I tell ordinary people that we're on the verge of solving one of the biggest open questions in all of biology?" Literally every single field was about to have an explosion of discoveries. All the textbooks would need to be completely rewritten. I'm glad to see this corollary was obvious to other biologists as well. :)
Second is that while I like the n-dimensional visual mappings of polysemantic signals to make the systems more interpretable/understandable as monosemantic simulations, that's probably only applicable to a subset of simpler polysemantic signals. Cell signaling will likely be largely resistant to that quick and easy visual explanation - even if the overall hypothesis ends up working out as expected and we're able to build monosemantic models from signals in superposition.
Third is that we haven't confirmed this hypothesis yet. Yes it's an obvious point, but as any bench scientist will tell you, no matter how enticing the hypothesis it'll probably still fail when it hits the lab. So I'm reserving some of my excitement for the possibility that this won't be validated in experiments. I mean, "it just HAS to work this way!" has never been a persuasive argument to my own experimental results.
Meanwhile, some thoughts on your other points from the post:
The biggest problem with the screening approach to drug discovery is that (as you know) we usually use cells in vitro or animal models during early drug discovery. Cells are preferred because they're cheap, but both these approaches struggle in many indications to generalize to complex human diseases such as psoriasis, RA, anxiety, and depression to name a few. It's notable that these fields tend to be very resistant to drug discovery compared to fields like virology, where we can easily dump something into a 384-well dish and see what happens. Whenever we run into something more complex than that, we struggle with the impossible task of creating models with the ability to translate into the clinic. A big part of the problem is that you can't know if something treats a complex disease unless you have most/all the elements of that disease in place interacting. The network effects aren't just at the level of molecular/cellular signaling. They're at the systems level, too, and likely also extend to microenvironment and temporal/pulse-based signals.
Biological systems aren't organized well for 'research and reductive understanding'; they're organized to integrate inputs efficiently and output robust signals for genetic survival. That means layers of complexity, and potentially LAYERS OF SUPERPOSITION. In other words, our AI models might be the *simplistic* version of the principle our biological mechanisms organize around. Virtual models that can parse monosemanticity from polysemantic signals might work for 'simple' AI, but we might need new tools to parse signals that are more developed than that.
Even so, there are probably SOME 'simple' polysemantic signals we'll be able to parse with this first generation of techniques, just as there were some direct signals we discovered with the old signal-response reductionist approach to cell signaling. I just hope we don't get bogged down in the new theory in the same way we were with the old/still-current theory.
Perhaps we already have a mechanism to prevent that, though? It will be exciting in the offing to see the emerging field of AI research seeding biological research, not just with novel tools to do the work, but also with novel hypotheses about mechanisms for storing/parsing complex network data - something we've ignored before because we had no way to approach the obvious open questions.
"It is these signals, not the proteins themselves, that ought to be targeted in drug discovery efforts." This is easier said than done. Indeed, it's difficult at the moment to articulate how we might approach this concept. We'll be MUCH closer, if this idea validates and we can shift the paradigm to start working out how to solve the polysemanticity problem, but I don't think we're even close to the point now where we can start promising medical innovations based on this new understanding. The earliest 'fruits' of this research might be if they're able to reliably predict intolerable adverse effects of prospective therapies, thereby weeding out many therapies that fail based on tolerability concerns. That would have the unintended consequence of boosting the number of false-positive therapies that make it to approval (purely from the statistical probability of shots-on-goal), but at least it should make the drug development process cheaper? Long-term, hopefully we can actually design from fundamental principles, as you suggest. We just need to figure out how that could be done.
"These issues are worsened by the fact that modern medical research has been building these networks over the past 80 years one edge at a time, using old techniques that modern science has largely proven to be inaccurate." This is the fundamental problem with the large corpus of knowledge accumulated in molecular/cell/signaling biology over the past three decades. It's all reported as "on/off" signals, but if what really matters is the DEGREE of activation somewhere between 0 and 1, then the last thirty years of work has only produced a partial list of the possible players in the game - if that! Since a system can as easily be coded to integrate points such as: X=0.8, Y=0.5, Z=0.1, signaling pathways once thought to be uninvolved might contrarily need to be essentially 'off' in order to transmit the signal in superposition. We may not be able to use much of ANYTHING specific from the past 30 years of cell and systems signaling research, when we map out the new signaling landscape under this new approach.
The multiplex assays will almost certainly help fill many of these gaps, but we'll have to go back and dig up the raw data (assuming it has been preserved after all these years, and not just converted into colorful/meaningless partially-labeled heat maps in papers people routinely glance over) before any of it will become useful enough to feed into an AI model that can parse the data. The more straightforward/reliable approach will be to just repeat the last 30 years of research, rebuilding everything from scratch.
Maybe that's depressing, but at least we spent the last few decades building out a lot of great new tools! So hopefully that speeds things up significantly.
Of course, we should all agree to continue ignoring post-transcriptional modifications as always because ... well if we're not sure how it applies it can't hurt us, right?