

Some Switches Can't Be Unflipped (But We Can Cut the Power)
Some Switches Can't Be Unflipped (But We Can Cut the Power)
Inherited mutations in growth genes, not just anti-growth genes, are found in cancer syndromes. The RET gene is an interesting example associated with many clinical conditions.

This article is part of a series on hereditary cancer syndromes and cancer genetics called Cancer Genomes. If new to the series, please go to my post “Introducing Cancer Genomes” for an explainer.
A 2014 review paper in Nature identified 114 genes relevant genes to inherited cancers. The Clinical Genome (ClinGen) Resource has rigorously evaluated this list confirming that there are almost 100 definitive gene-to-cancer associations.1 Of these, there are fewer than 10 classic proto-oncogenes.2 In the case of hereditary cancer, it is simply much more common to find the offending mutation in a tumor suppressor gene than an oncogene. This doesn’t directly speak to the actual incidence of cancer syndromes driven by mutations in tumor suppressor genes or oncogenes and the distinction is not as complete and neat as we like to imagine, but it is all but certainly the case that mutations in tumor suppressors are more common because there is weaker selection against them. The disease mechanism is typically dependent on the occurrence of a second hit so the first hit can be passed on without affecting offspring before reproductive maturity.
There are theoretical principles and empirical observations that support this conclusion. First, the mechanism by which an inherited mutation drives disease in the context of a tumor suppressor gene versus an oncogene clearly differ. Pathogenic variants in tumor suppressor genes tend to be loss-of-function in effect, while the opposite is true for oncogenes, i.e. gain-of-function variants. Second, the penetrance of disease in cases of variants in tumor suppressor genes is lower than that of oncogenes. Sometimes mutation carriers never get cancer at all! This again is because of the need for a second hit to the genome to kick off carcinogenesis, where the aberrant oncogene has a head start. It is hard to escape health issues altogether when inheriting a gain-of-function mutation in an oncogene.

Today, we’ll examine our first oncogene associated with hereditary cancer, RET. RET, an abbreviation meaning "rearranged during transfection,"3 encodes a receptor tyrosine kinase (RTK). RTKs are membrane-spanning proteins that conduct external inputs to the interior of cells. The input, often a small protein - often called a ligand - floating in the interstitial fluid outside a cell, typically induces the RTK to add a phosphate groups to itself, a process aptly called autophosphorylation.4 RTKs are often proto-oncogenes because they are situated at the top of a signaling cascade and signaling cascade tend to coordinate cellular behaviors that if left unchecked drive cancer processes: growth, proliferation, survival, etc. Also, there are a number of molecular changes that can lead them to become abnormally activated, such as gain-of-function mutation, gene amplification, gene rearrangement, and overexpression. Plus, kinases are often the positive effectors of signaling pathways while phosphatase are negative effectors. So if your in a pinch in a cancer biology class and don’t know anything except whether a gene is a kinase or phosphatase, guess the former is an oncogene and the latter is a tumor suppressor.5

Inherited mutations in RET are associated with a cancer syndrome called Multiple Endocrine Neoplasia Type 2 (MEN2). MEN2 comes in two varieties, A and B.6 Both involve an increased risk for a type of neuroendocrine tumor called pheochromocytoma that irregularly secretes epinephrine and norepinephrine. Pheochromocytoma grow from the chromaffin cells of the adrenal gland as suggested by the name. The A type or MEN2A is characterized by an increased risk for parathyroid adenoma or hyperplasia, while MEN2B is characterized by medullary thyroid cancer (MTC) along with a suite of other features: lesions on the lips and tongue, distinctive facial features like enlarged lips, hamartomas of the GI tract, and a Marfan syndrome-like builds.

RET is derived from the strange phrase "rearranged during transfection” because it was first identified in a switched around (rearranged) region of a chromosome and that switching around was observed during a transfection experiment. A transfection is a type of cell culture experiment that introduces some arbitrary, pre-determined amount of foreign DNA into a putative host cell. The goal and results of these experiments can vary substantially. In this case, DNA from human lymphoma cells was introduced into mouse fibroblasts, which are a type of connective tissue cell used commonly in cell culture work. Then, the transfected cells were assessed via a cloning and blot hybridization approach, which found that a genetic region was rearranged during the transfection. And thus, RET was born. It was also clear that RET was functioning to spur abnormal growth of cells. In other words, it was an oncogene.
A few years later, fluorescence in situ hybridization studies7 localized RET to a portion of the long arm of chromosome 10, 10q11.2.8 This work also asserted that RET made a reasonable candidate for MEN2A. This was confirmed by a series of studies in 1993 that successfully linked the RET gene region and then mutations in the RET gene to MEN2A and medullary thyroid cancer cases.9 Additional research in 1993, mapped the structure of the RET gene, defining 20 total exons and the presence of alternative spliced transcript products.
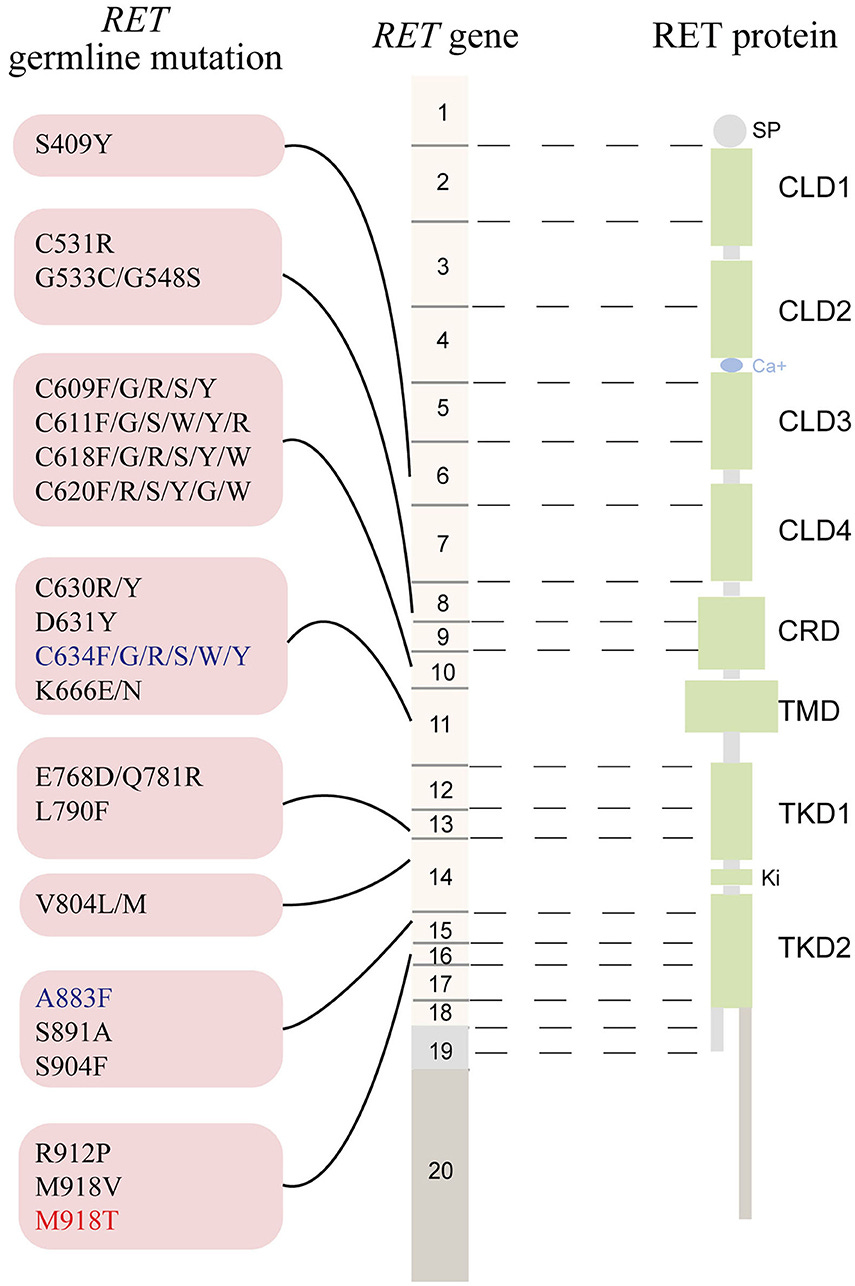
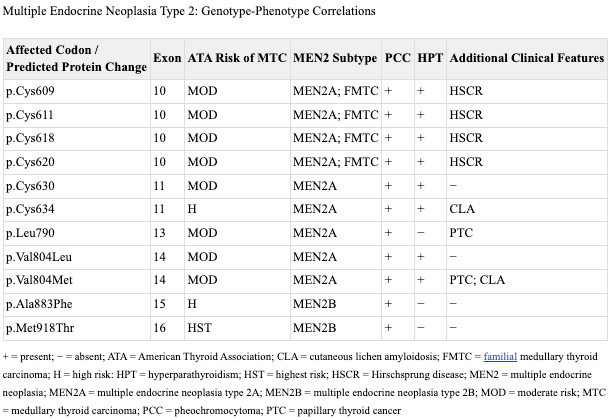
At this moment, to the extent there was any existing confusion about RET, it concerned the number of phenotypes linked to the gene. Not only was MEN2A genetically linked but so was MEN2B, familial medullary thyroid cancer (FMTC), and Hirschsprung disease (HSCR).10 This morass of genotype-phenotype relationships required a great deal of research to sort out.
The short story is that the cancer phenotypes (MEN2 & FMTC) are associated with heterozygous gain-of-function mutations that cluster in specific regions of RET, exons 10, 11, 13, 14, 15, and 16, while heterozygous loss-of-function mutation are associated with HSCR and are observed throughout the gene. This breakdown is true for a lion’s share of cases of HSCR (~50% of familial cases0, but there are also cases where there will be a confluence of phenotypes that segregate with the same RET mutation. This is largely specific to mutation involving the cysteine codons 609, 611, 618, and 620 in exon 10.
The Bright Side of Oncogene Mutations
Now, it is never fair to characterize the inheritance of rare disease-causing mutations as positive, but the silver lining when dealing with mutations in oncogenes, especially kinases, is ease of druggability. It is a simple proposition: find a small molecule with drug-like properties that can stick itself into the oncogene structure and stop its activity. RET is exactly this sort of straightforward situation and drugging RET, whether it is driving a familial or sporadic cancer, has been shown to be dramatically effective in many cases.

Today, there are 4 approved drugs with activity against RET: vandetanib, cabozantinib, pralsetinib, and selpercatinib. They’re a mainstay of treating medullary thyroid cancer (MTC), improving survival outcomes.11 Vandetanib and cabozantinib are quite non-specific tyrosine kinase inhibitors (TKI) that affect many kinases. Pralsetinib and selpercatinib are selective inhibitors specific to RET. Subsequently, their use is restricted to patients with RET alterations, which makes the use of molecular diagnostic tools of high importance in any MTC case. In fact, selpercatinib actually carries a pan-tumor approval so that a patient of any solid cancer type can receive selpercatinib if they have an activating mutation in RET.
The path from the initial genetic discovery to an approved drug was less than 20 years, which considering the historical context is fairly impressive. The first drug in the same class, TKI, was approved in 2001, imatinib, and that was in a blood cancer. Historically, the developmental life cycle for any given drug from pre-clinical work to clinical research to market authorization averages about two decades anyway. Although the RET inhibition development story isn’t entirely one of rational design,12 quite a bit of serendipity was involved, it is nonetheless an exemplary case of genetic data identifying a clear mechanism to drug and targeting that mechanism has led to bonafide clinical benefit. So the 1993-to-2011 timeline is artificially extended and hence the road to even more selective RET inhibitors has been faster.
The study of a familial condition, MEN2 and family thyroid cancers, led to a genetic discovery, RET, that inspired pharmacological innovation that is now saving lives. This importantly illustrates that genetic knowledge has clinical utility beyond diagnosis and prognosis. It can affect every aspect of clinical management.
Currently, ClinGen has 102 gene-disease curations at definitive, 3 at moderate, 3 at limited, 1 at disputed, and 6 at refuted classifications. It is important to note that a gene can have more than one disease association. The full list of ClinGen’s gene-disease validity curation can be accessed here.
This list excludes RASopathy genes. These are evaluated by a separate cancer panel. Subsequently, it isn’t a comprehensive list. However, this doesn’t detract from the overall point here, which is the TSGs outnumber proto-oncogenes in terms of raw number of associations with cancer syndromes.
We’ll touch on why this is the name soon.
This is a bit of a simplification. Sometimes an RTK adds the phosphate to itself, while other time it add it to a nearby version of itself.
There are of course exceptions to this. For example, PTPN11 encoding SHP2 is an phosphatase that acts like an oncogene.
My PhD advisor Dr. Charis Eng is an expert in MEN2 and hence author the GeneReviews page.
An approach covered in “The Tumor Suppressor to Rule Them All”
Remember the first gene in the Cancer Genomes series was PTEN and is also on the long arm of chromosome 10 and when mutated shares some phenotypic overlap with MEN2A/B! For whatever reason, the q arm of chromosome 10 seems really critical to regulating cellular growth.
These studies are Lairmore et al. (1993), Mulligan et al. (1993), and Donis-Keller et al. (1993).
Hirschsprung's disease is a birth defect affecting the large intestine via deficits in nervous tissue. It causes issues with passing stool.
For metastatic MTC treated with selpercatinib, the overall five-year disease-specific survival is about 50%, which is about double the base survival rate in this patient group.
The first RET inhibitor to be approved, vandetanib, was only found to have activity against RET in 2002, only 9 years before its approval.